
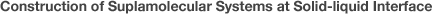
Scanning tunneling microscopy (STM) was invented in 1982, and has allowed significant advances in various scientific fields through facilitating the real space imaging of molecules on surfaces. Molecular imaging elucidating the arrangements in arrays, orientations and even intramolecular structures has been achieved in air, ultrahigh vacuum (UHV) and solution. Construction and in-space visualization of supramolecular adlayers on well-defined single crystal surfaces have received significant attention in the last decade. Coupling supramolecular chemistry and scanning probe microscopy allows the visual consideration of molecular self-assembly in sub-molecular space.
The adlayer structures of organic molecules are generally controlled by the balance between adsorbate-substrate (epitaxial) interactions and intermolecular ones. Molecular adsorption on well-defined single crystal surfaces has often been considered from a static crystallographic point of view, i.e., simply as a matter of surface chemistry. In such systems (known as epitaxial adsorption or epitaxy) predominantly static structures have been discussed with reference to properties like adlayer commensuracy and adsorbate orientation. Conversely, the self-assembly of sophisticated and highly ordered supramolecular adlayers has been predominantly discussed in terms of intermolecular interactions rather than epitaxial arrangements in surface chemistry. In the case of weak or mild adsorption (typically physisorption), molecules on surfaces are relatively unrestrained by the substrate lattice, thus isolated molecules on surface can move spontaneously to reach a thermodynamically stable adlayer structure. In other words, artistic 2D-supramolecular systems are regulated by intermolecular interactions and subsequent molecular motion in weak adsorption systems.1-5
Control of the adsorption strength is one of the most crucial points for constructing highly-ordered adlayers. Isothermal adsorption at the solid–liquid interface can be classified into four states related to adsorption strength (adsorbate–substrate interaction), as shown in Figure 1. The condition indicated by light blue in Figure1 is where highly ordered molecular adlayers are constructed, and this is based on intermolecular interactions rather than interaction with the substrate. In these thermodynamically controlled relatively weak adsorption conditions, the adsorption–desorption equilibrium and lateral diffusion of molecules on the surface are highly maintained. Self-organization at solid–liquid interfaces is then achieved by intermolecular interactions as the driving force of 2-D crystallization.
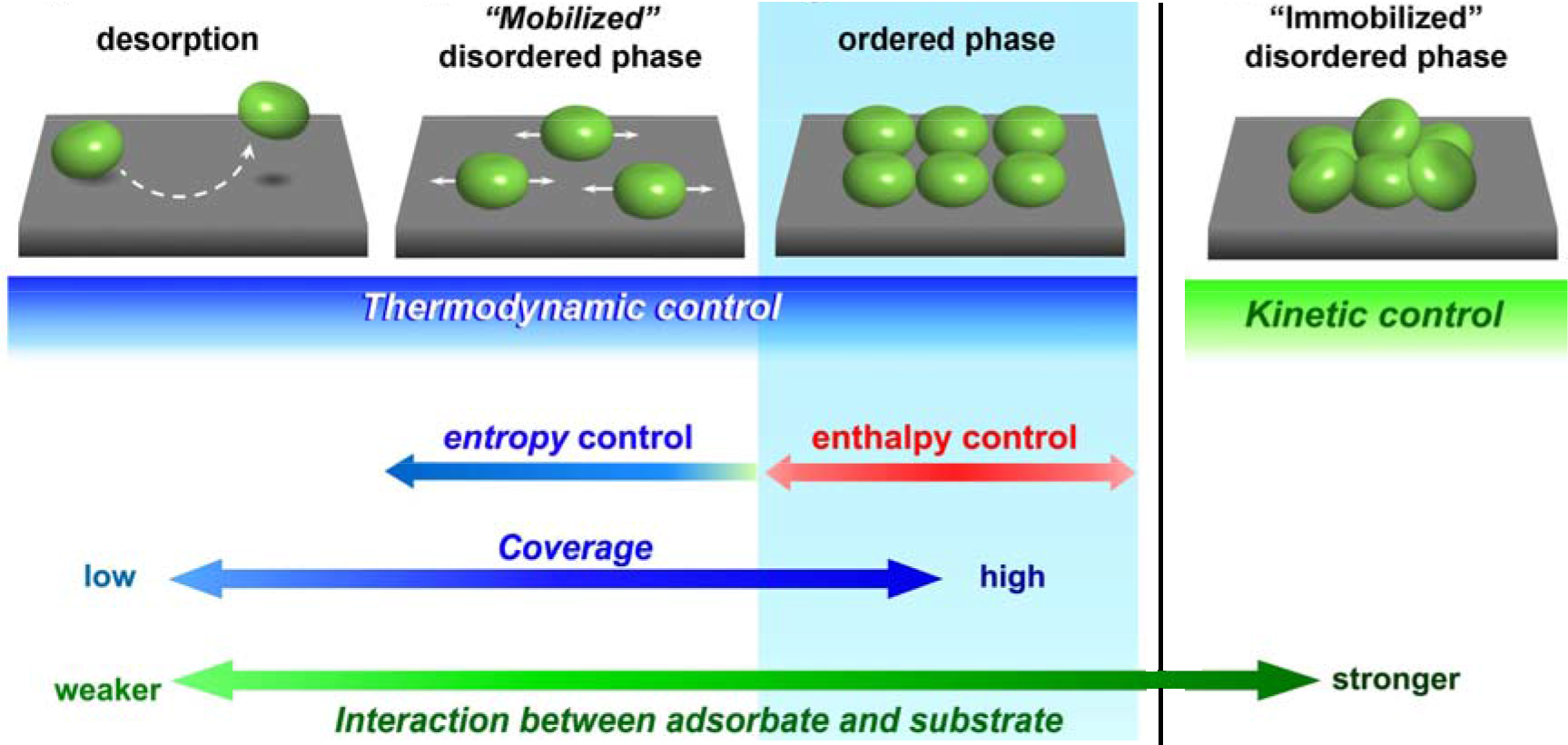 Figure1Schematic representation of molecular structure on surface at various strength of substrate-molecular interaction.
Figure1Schematic representation of molecular structure on surface at various strength of substrate-molecular interaction.
We have reported the 2D self-assembly of trimesic acid (TMA) driven by their intermolecular hydrogen bond.1 TMA is known to form a network structure by forming intermolecular H-bonds between carboxylic acid residues in bulk crystals. Figure 2 (Left) shows typical STM images of the "2D-network" of the TMA adlayers. The "2D-network" array extended over the wide and atomically flat terrace of Au(111). Despite the relatively large area covered by the image, the individual features of the "2D-network" can be recognized. The uniformity of the structure is quite high, and there are very few phase boundaries. Interestingly, these "2D-network" arrays were typically observed on the reconstructed Au(111) surface.
Based on a similar fashion, we have revealed that melem, which is a condensed melamine derivative, can also form the H-bond driven 2-D self-assembled structure at the interface between aqueous solution and Au(111).2 Melem and its polymeric derivatives have attracted attention because of properties such as photocatalysis and fluorescence with rare metals. Melem has potential as a building block for supramolecular architectures because of these features. Throughout the electrochemical investigations and in-situ STM observations at the various concentrations, polymorphism of the self-assembled structures was controlled by the concentration and electrochemical potential control. (Figure 2 Right)
 Figure2The hydrogen-bond-driven 2-D pattern structures formed from trimesic acid (TMA) and melem.
Figure2The hydrogen-bond-driven 2-D pattern structures formed from trimesic acid (TMA) and melem.
Recently, we have reported a general substrate-mediated, soft solution methodology for the preparation of extended covalently interconnected polymeric nanoarchitectures in low-dimensions.3-5 Substrate supported systems are commonly self-produced through relatively weak noncovalent interactions such as van der Waals interactions, hydrogen bonds, and π-π stacking. As expected, it has proven difficult to avoid unfavorable irreversible cross-linking which leads to formation of disordered 2- or 3-D nanostructures in substrate supported, covalently bonded molecular systems. To overcome this issue, we focused on equilibrium polycondensation reactions to connect building blocks in extended frameworks. The reversibility and thermodynamic control of this solution reaction encouraged us to extend the conventional process of molecular self-assembly in 2-D from noncovalently bonded systems to covalently bonded ones.
The STM image revealed the framework structures to be composed of 4-fold amine molecules interconnected by covalently bonded linker molecules (Figure 3). The frameworks, with their corresponding structural variations, extended over wide areas and demonstrated strong agreement with the corresponding model predictions. The surface-mediated, solution-based synthetic methodology presented here introduces the unique concept of utilizing thermodynamic control of an equilibrium polymerization reaction toward preparation of robust, extended supramacromolecular networks and arrays. This methodology eliminates the necessity for severe conditions and sophisticated equipment common to most current fabrication techniques and imparts almost infinite possibilities for the preparation of robust materials with designer molecular architectures.
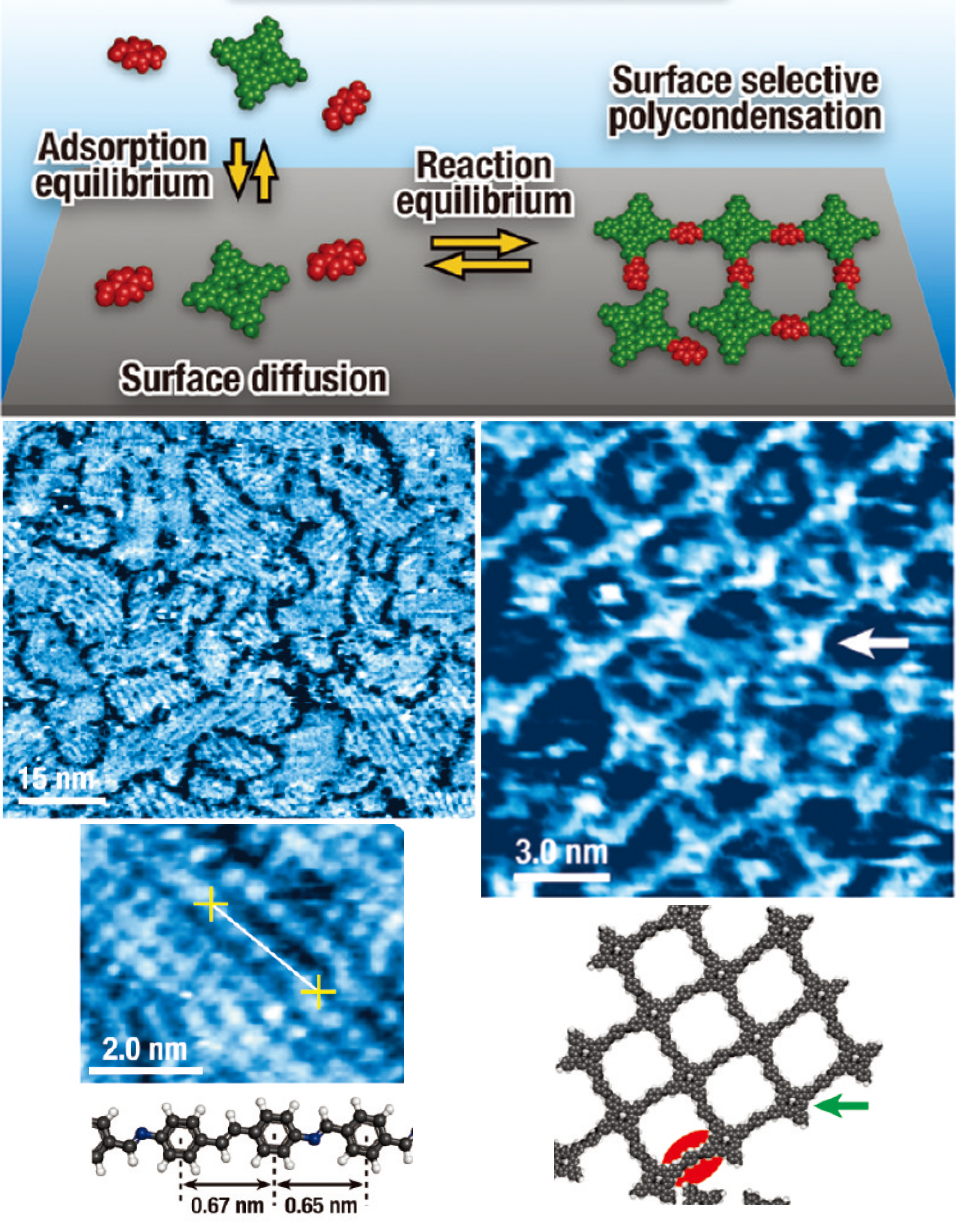 Figure3The schematic representation of self-assembly of covalent-bonded nanostructures by spontaneous equilibrium reaction on solid-liquid interface. STM image of self-assembled 1-D linear polymer and 2-D frameworks.
Figure3The schematic representation of self-assembly of covalent-bonded nanostructures by spontaneous equilibrium reaction on solid-liquid interface. STM image of self-assembled 1-D linear polymer and 2-D frameworks.
π-Conjugated polymer thin films will be attractive for applications as organic semiconductor materials such as light-emitting diodes, solar cells, and field-effect transistors. Preparation of thin films of the polymers on an arbitrary surface is crucial for assembling the electronic devices. We have tried to extend the methodology to obtain self-construction of 2-D covalently bonded nanoarchitectures to continuous 3-D growth of similar polymer systems (chemical liquid deposition; CLD).6-7
Colored Schiff base π-conjugated polymer thin films was prepared by having solution pH slightly higher than that for monolayer systems, which is because the reaction equilibrium was slightly forwarded. The polymer films spontaneously form under ambient conditions by simple immersion of graphite substrates in an aqueous solution containing the monomer units. CLD is achieved by delicate control of solution pH, which allows surface selective polymerization and deposition but inhibits reaction in the aqueous phase. Colored π-conjugated polymer films can be prepared on highly oriented pyrolytic graphite by simple immersion in an aqueous solution containing aromatic amines and aldehydes under delicate pH control. (Figure4)
This CLD method allows the preparation of designable π-conjugated polymer films by simple immersion of a substrate in an aqueous solution containing the monomer building blocks. Tuning of the optical and electrical properties of the polymer films and the introduction of functional groups could be easily obtained by the selection of the functional building blocks.
 Figure4Appearance of poly(azomethine) films deposited on a HOPG with varying deposition time, and AFM and SEM images of organic polymer thin film formed by CLD.
Figure4Appearance of poly(azomethine) films deposited on a HOPG with varying deposition time, and AFM and SEM images of organic polymer thin film formed by CLD.
Copyright © 2008- KUNITAKE Laboratory.
